Research
My research focuses on developing and applying advanced computational methods to understand the electronic and optical properties of emerging quantum materials. This involves both method development and their application to specific material systems like 2D materials, nanostructures, and molecular assemblies.
Semi-Empirical Pseudopotential Method (SEPM)
A significant part of my work involves the development and application of the Semi-Empirical Pseudopotential Method (SEPM). This method provides an efficient and accurate way to calculate the electronic structures of large and complex material systems. Our SEPM approach typically combines two-dimensional plane waves with B-spline functions for the perpendicular direction, and incorporates both local and non-local potential terms. These terms are carefully parameterized to reproduce key quantities obtained from first-principles Density Functional Theory (DFT) calculations or experimental data. This work is often in collaboration with Prof. Chung-Yuan Ren (NKNU).
Applications to Graphene and Graphene Nanoribbons
We have successfully applied SEPM to investigate the electronic properties of graphene and various armchair graphene nanoribbons (aGNRs). The method accurately captures the characteristic Dirac cones in graphene and the band gap variations in aGNRs depending on their width and edge structure. This allows for efficient exploration of these materials for potential nanoelectronic applications. Details can be found in our publication: Semi-Empirical Pseudopotential Method for Graphene and Graphene Nanoribbons (MDPI Nanomaterials 2023).
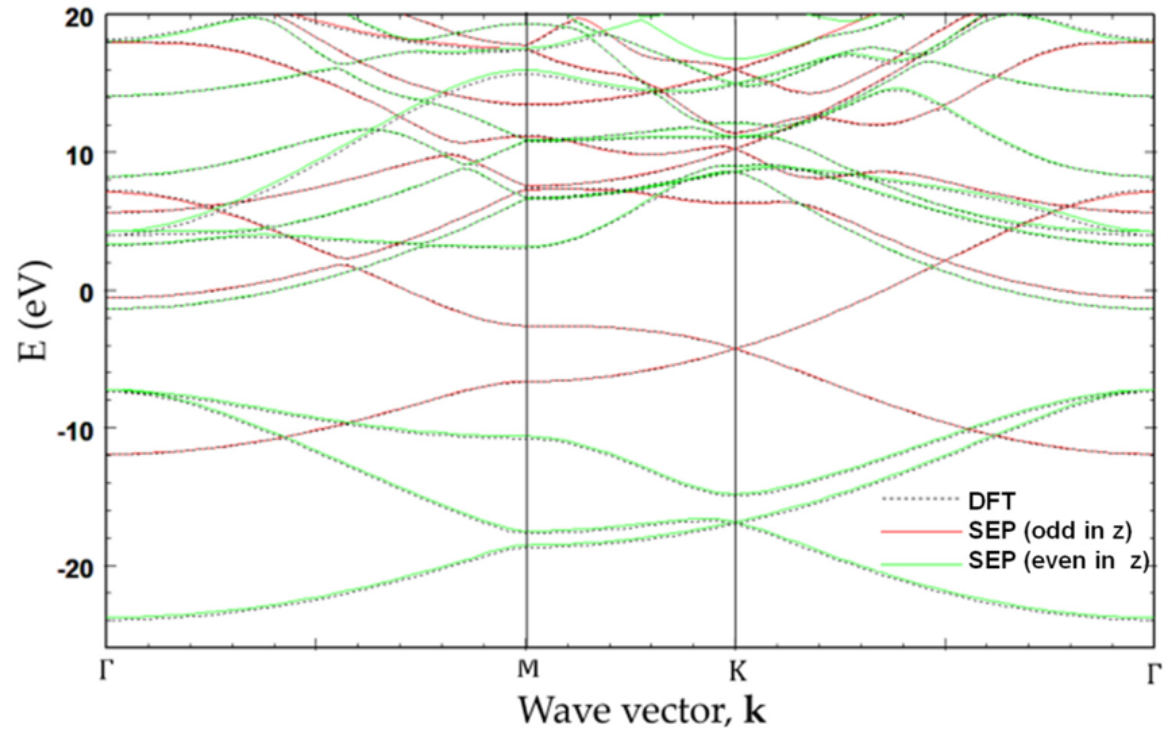
Fig 1: Comparison of graphene band structure via SEPM (solid) and DFT (dashed).
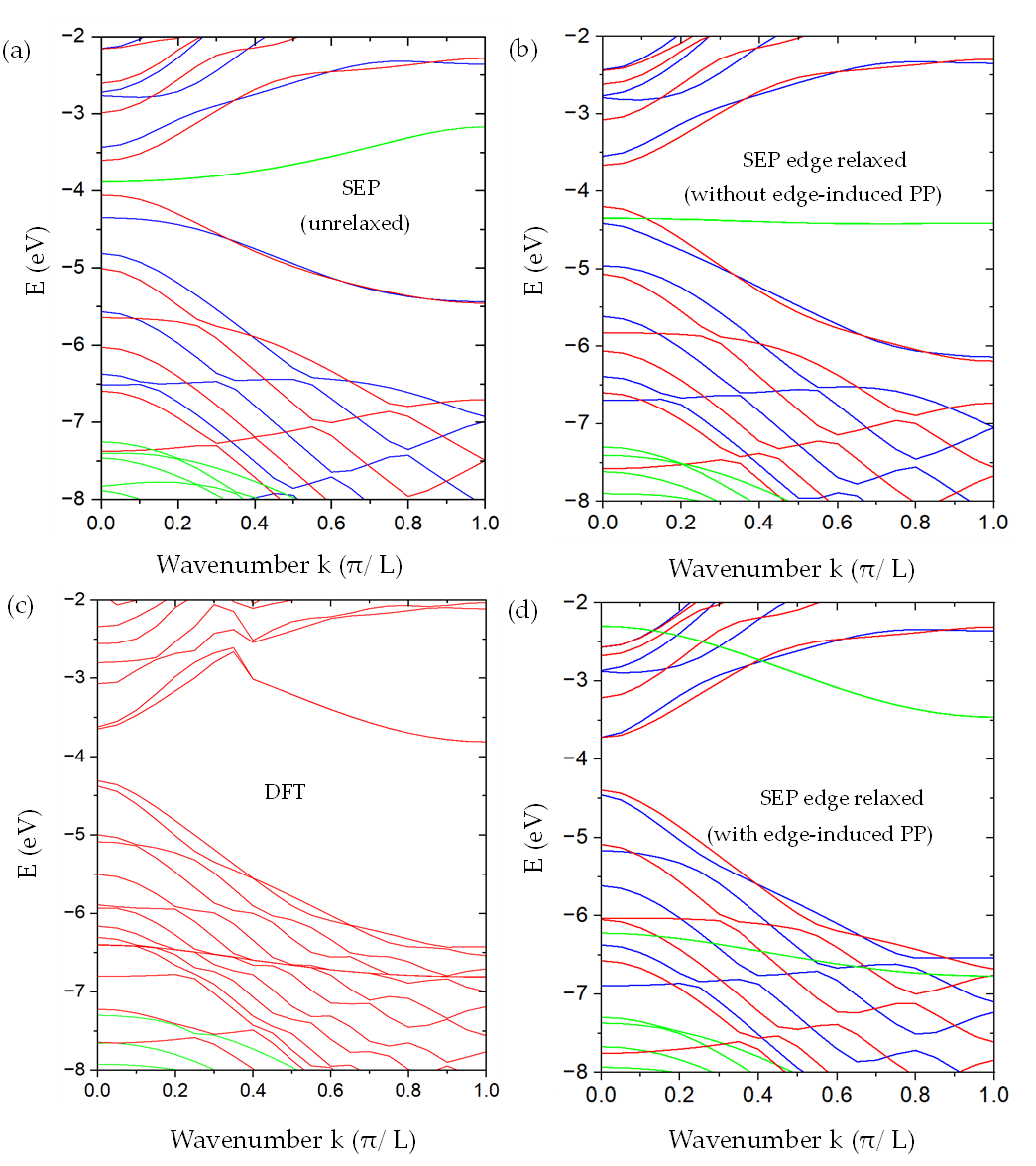
Fig 2: Comparison of aGNR band structures from DFT (dashed) and SEPM (solid).
Applications to Layered Transition-Metal Dichalcogenides (TMDCs)
The SEPM framework has also been developed for monolayer Transition-Metal Dichalcogenides (TMDCs). This allows for accurate and efficient calculation of their electronic band structures, which is crucial for understanding their optical and transport properties, including exciton formation. Our work aims to provide a robust tool for exploring various TMDCs and their heterostructures. Further details are available in our preprint: Semiempirical Pseudopotential Method for Transitional-Metal Dichalcogenides (arXiv:2406.15913).
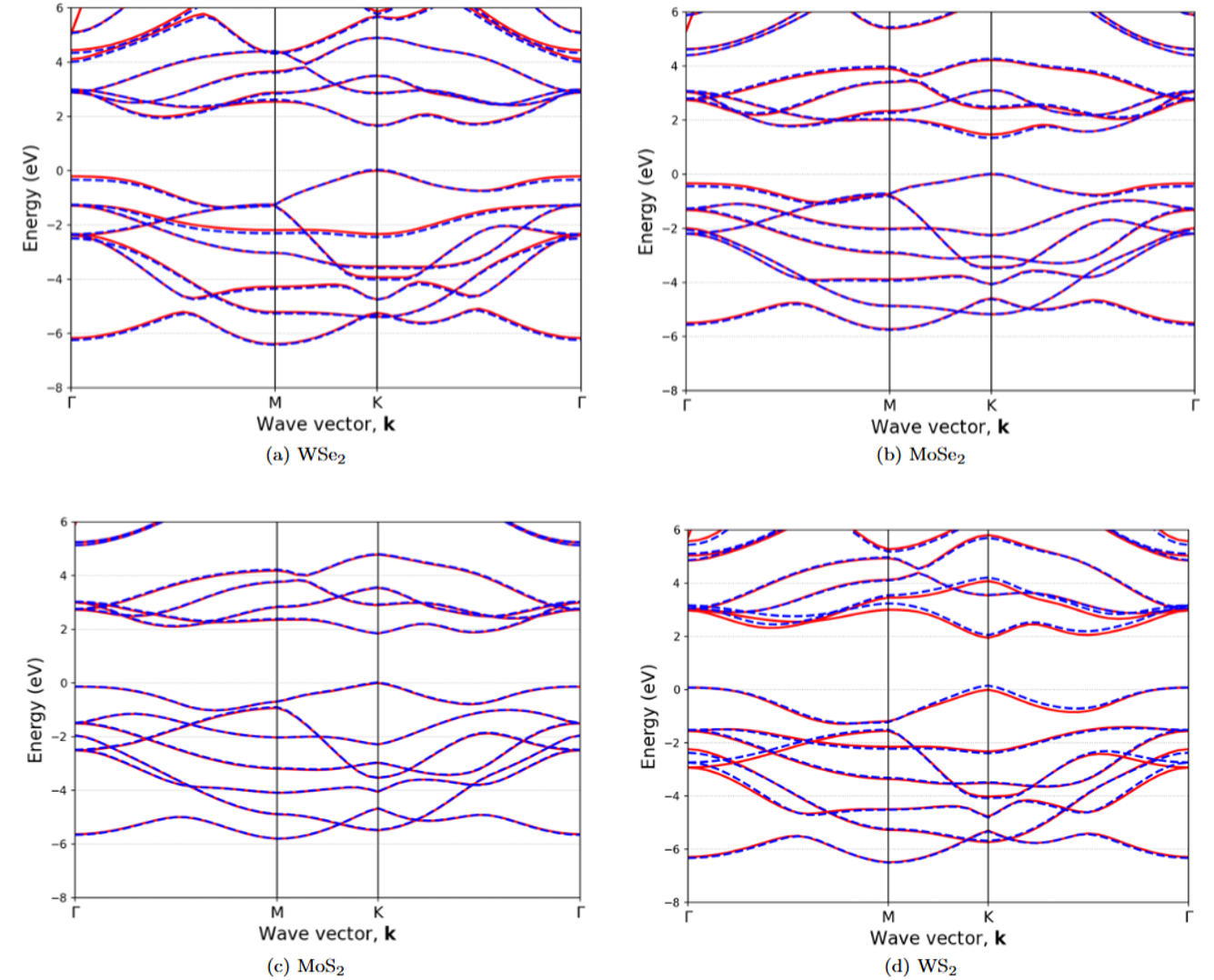
Fig 3: Band structure comparison for monolayer TMDCs: SEPM (solid lines) vs. DFT (circles).
Exciton Physics in Transition Metal Dichalcogenides (TMDCs)
Monolayer and few-layer Transition Metal Dichalcogenides (TMDCs) are fascinating materials exhibiting strong light-matter interactions dominated by excitons—bound electron-hole pairs. My research in this area focuses on developing theoretical frameworks and employing advanced many-body approaches to model exciton binding energies, their dynamics, and optical properties in monolayer and bilayer TMDCs. Understanding and controlling exciton behavior is key to harnessing TMDCs for optoelectronic devices like LEDs, solar cells, and valleytronic applications. Current projects include investigating 2P interlayer excitons and electric-field-driven brightening of interlayer excitons.
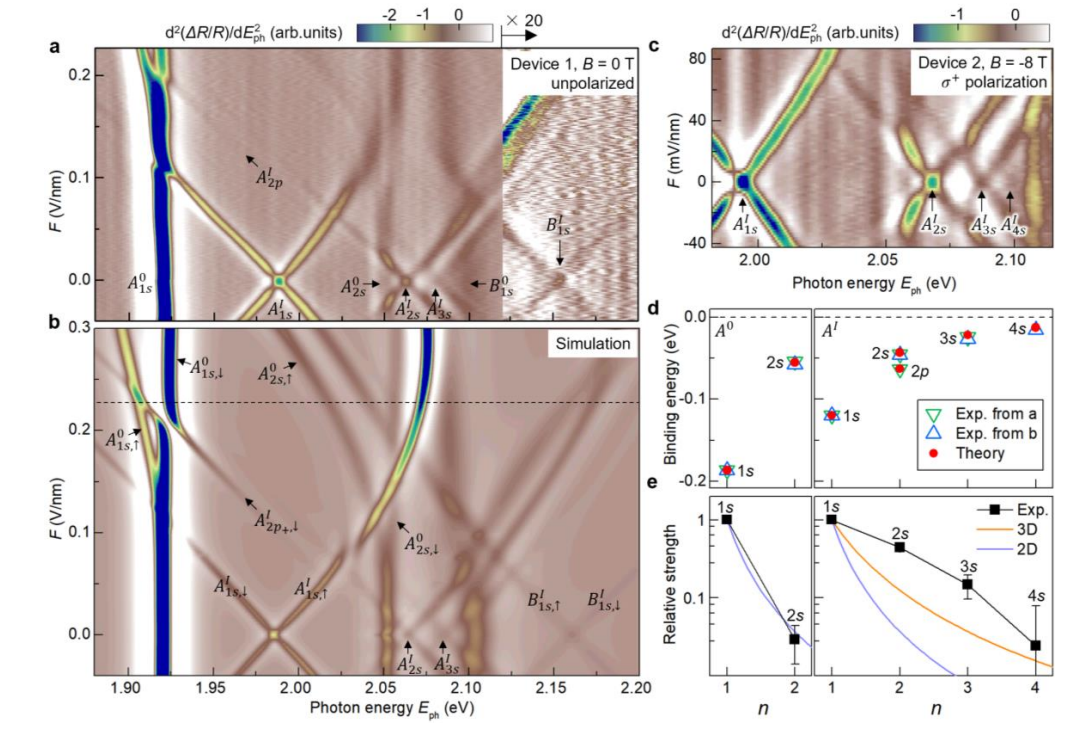
Fig 4: Calculated s-like and p-like interlayer exciton states in bilayer MoS₂.
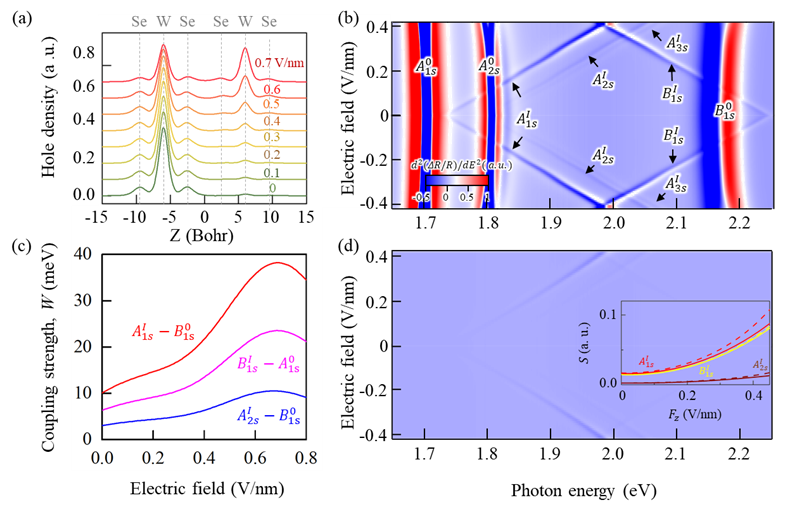
Fig 5: Electric field tunes hole distribution and exciton properties in bilayer WSe₂.
Future Directions
Future work aims to extend these computational models. One key goal is to use planar-basis Density Functional Theory (DFT) calculations and further refine SEPM to accurately model van der Waals (vdW) interactions within bilayers of graphene and TMDCs. The formation of moiré superlattices due to lattice mismatch in stacked TMDC layers leads to periodic variations in the electronic potential. By efficiently calculating the vdW-induced moiré superlattice potential, we can gain valuable insights into the band-edge profiles at different twist angles and explore the intriguing electronic phenomena in these systems, such as moiré excitons and correlated electronic states.